Estimating effect sizes with a DeepDive Model#
This tutorial demonstrates how to estimate effect sizes with a DeepDive model on single-cell ATAC-seq data.
Imports#
[1]:
import scanpy as sc
import DeepDive
import seaborn as sns
import numpy as np
import matplotlib.pyplot as plt
[2]:
from utils import reads_to_fragments
1. Load and preprocess the dataset#
We start with an AnnData object containing single-cell chromatin accessibility profiles. Here we use the liver sciATAC-seq3 dataset (sciatac3_liver_10k.h5ad), subset from the full dataset (dataset and preprocessing better described in the training.ipynb notebook).
[3]:
adata = sc.read_h5ad('data/sciatac3_liver_10k.h5ad')
# Filter genes detected in fewer than 1% of cells
min_cells = int(adata.shape[0] * 0.01)
sc.pp.filter_genes(adata, min_cells=min_cells)
# Ensure unique cell IDs
adata.obs_names_make_unique()
# Convert raw reads into fragments
reads_to_fragments(adata)
adata.X = adata.layers['fragments']
2. Define model and training parameters#
We specify model hyperparameters and training settings.
[4]:
n_decoders = 5
model_params = {
'n_epochs_pretrain_ae' : 200*n_decoders,
'n_decoders' : n_decoders,
}
train_params = {
'max_epoch' : 300*n_decoders,
'batch_size' : 1024,
'shuffle' : True
}
[5]:
discrete_covriate_keys = ['sample_name', 'sex', 'batch', 'cell_type']
continuous_covriate_keys = ['day_of_pregnancy']
3. Train DeepDive#
We initialize and train the model using the training dataset.
[6]:
model = DeepDive.DeepDive(adata = adata,
discrete_covariate_names = discrete_covriate_keys,
continuous_covariate_names = continuous_covriate_keys,
**model_params
)
[7]:
model.train_model(adata, None,
**train_params)
Epoch Train [1500 / 1500]: 100%|██████████| 10/10 [00:00<00:00, 10.10it/s, ETA=01d:00h:21:m41s|01d:00h:21:m41s, kl_loss=1.68, recon_loss=3.15e+3]
4. Compare groups#
The DeepDive.compare_groups() function identifies accessible regions that differ between two groups independent of disentangled covariates.
In this example, we compare Hepatoblasts and Erythroblasts to identify differential chromatin accessibility patterns.
[8]:
da = DeepDive.compare_groups(model, adata, 'cell_type', groupA = 'Hepatoblasts', groupB = 'Erythroblasts')
[9]:
da.head()
[9]:
| Feature | groupA | groupB | Difference | Zscore | Pvalue | FDR | |
|---|---|---|---|---|---|---|---|
| 0 | chr1:565561-565842 | 0.808578 | -1.587507 | 2.396085 | 1.704382 | 0.006697 | 0.055971 |
| 1 | chr1:569259-569557 | 2.841677 | 1.127797 | 1.713880 | 1.223938 | 0.128054 | 0.535595 |
| 2 | chr1:569659-569951 | 1.822991 | -0.457494 | 2.280485 | 1.626589 | 0.011054 | 0.082341 |
| 3 | chr1:713647-714744 | 0.733735 | 0.902667 | -0.168932 | -0.094762 | 0.999939 | 1.000000 |
| 4 | chr1:762075-763248 | -0.183651 | -0.042263 | -0.141388 | -0.077697 | 0.999797 | 1.000000 |
This returns a DataFrame containing:
Difference: Effect size between the two groupsFDR: False discovery rate (multiple testing–corrected p-value)
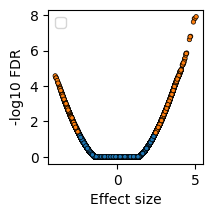
We can visualize the differential results as a volcano plot.
[10]:
plt.subplots(figsize=(2,2))
sns.scatterplot(
x=da.Difference,
y=-np.log10(da.FDR),
hue=da.FDR < 0.05,
edgecolor='k',
linewidth=0.5,
s=10
)
plt.legend([])
plt.ylabel('-log10 FDR')
plt.xlabel('Effect size')
[10]:
Text(0.5, 0, 'Effect size')
5. Compare groups with covariate constraints#
DeepDive can also control for confounding factors by fixing certain covariates. For example, to compare Hepatoblasts vs Erythroblasts while holding sex = female constant:
[11]:
background = {'sex':'female'}
da = DeepDive.compare_groups(model, adata, 'cell_type', groupA = 'Hepatoblasts', groupB = 'Erythroblasts', background = background)
This performs the same comparison as above but within the context of a fixed background covariate — allowing interpretation of group differences dependent the sex.
Summary#
DeepDive.compare_groups()quantifies differential effects between categorical groups.Supports covariate-controlled comparisons via the background argument.
Returns a feature-level DataFrame with effect sizes and FDR-adjusted p-values.
[ ]: